医药品医疗器械等法的概要
更新日:2024年3月20日
通过从药事法到医药品医疗器械等法的修改,以确保医药品、医疗器械等的安全和迅速供应为目的,创设了附件文书的备案义务,强化了医疗器械的注册认证机构的认证范围的扩大等的安全措施,构筑了与医疗器械和体外诊断用药品特性相适应的监管框架。
医疗器械相关的主要修改之处
医疗器械制造业相关变更
(1)制造业从许可制或认定制变更为注册制
在过去,日本国内制造业是许可制度,而外国制造商是认定制度,现在两者都更改为注册制,并且要件也得到了简化。过去的制造分类不复存在,制造业的应注册范围(对象)也发生了变化。
| 修改前 | 修改后 | |
| 许可/注册等 | 日本国内:许可、外国:认定 | 日本国内/外国:注册 |
| 有効期间 | 5年 | 5年 |
| 许可/注册等权力主体 | 日本国内:都道府県、外国:国 | 日本国内:都道府県、外国:国 |
| 制造分类 | 一般、灭菌、生物、包装等 | 无 |
| 许可/注册等的要件 | 不适格条件 | 不适格条件 |
| 结构设备要求 (一般/灭菌/生物/包装等分类各有要求) | 无(必要事项由新QMS省令规定) | |
| 责任技术者 | 必要 | 必要 |
因为已经不对制造进行一般和包装等的分类,药事法中作为许可要件的药局等结构设备规则也不再适用。
(2)需要注册的工程的变更
以前不是许可等的对象的进行设计的设施变成了需要注册的对象,“软件”以及“软件记录媒介”也作为医疗器械需要进行注册。
| 制造工程 | ①医疗器械 (②③④以外) | ②一般医疗器械 | ③单独的软件 | ④单独的记录软件的媒介 |
| 设计 | ○ | × | ○ | ○ |
| 主要组装 | ○ | ○ | × | × |
| 灭菌 | ○ | ○ | × | × |
| 在日本国内进行最终制品的保管 | ○ | ○ | × | ○ |

医疗器械的定义相关变更
在药事法规制范围之外的”软件”本身、已经变为与国际接轨的新法律的规制对象,“软件”及“记录软件的媒介”已被追加入医疗器械的定义中。
属于医疗器械的软件的范围如下。
软件
1.用于疾病诊断的软件
2.用于疾病治疗的软件
3.用于疾病预防的软件
记录软件的媒介
1.记录用于疾病诊断的软件的媒介
2.记录用于疾病治疗的软件的媒介
3.记录用于疾病预防的软件的媒介
医疗器械制造贩卖业的许可要求、遵守事项的审查等(QMS相关等)。
QMS制度的重组
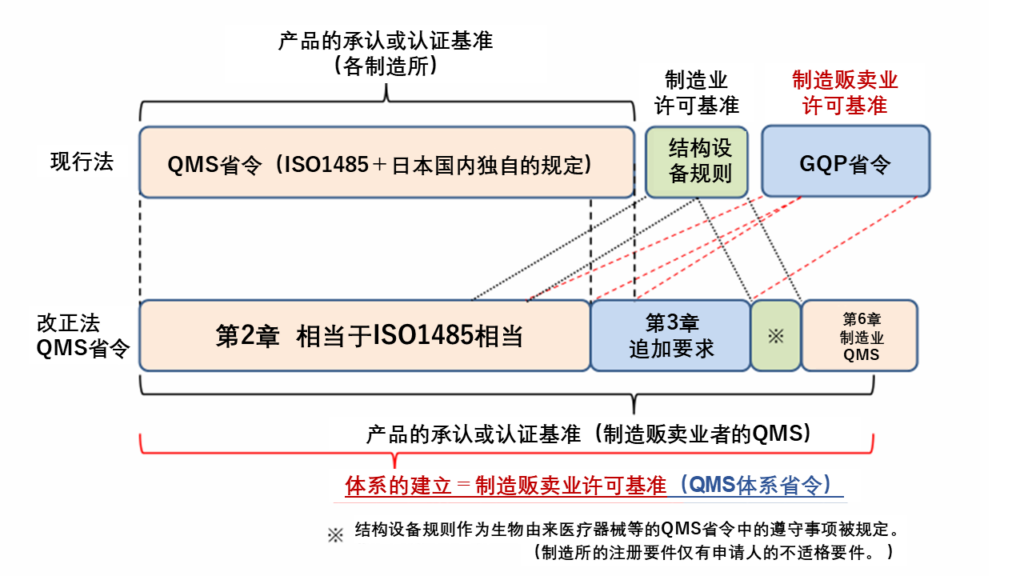
(1)QMS省令已经修改,并适用于制造贩卖业。
过去,对于医疗器械制造贩卖业者,要求基于GQP和GVP品质管理手册的记录,但现在GQP已被QMS取代,需要建立一个涵盖相关制造业者的整体品质管理监督系统基准书(品质管理手册)。
(2)符合取代GQP省令的QMS体制省令已变成医疗器械等的制造贩卖业的许可要件。
《与进行医疗器械或体外诊断用医药品的制造管理或质量管理业务的体制的基准相关的省令》(平成26年8月8日厚生劳动省令第94号)也适用于仅经营一般医疗器械的制造销售业者和制造业者。
(3)产品群调查的合理化
产品群省令(规定了关于确保医药品、医疗器械等的品质、有效性和安全性的法律第23条之2之5第7款第1号所规定的医疗器械或体外诊断用药品的分类的省令)”已经被制定,对于同一产品群或同一注册制造所的产品,不再需要进行调查。
医疗器械认证制度的相关变更
(1)注册认证机构进行第三方认证的范围已经扩大
不仅是管理医疗器械,甚至包括厚生劳动大臣制定基准指定的高度管理医疗也变成民间的注册认证机构的认证对象。
(2)在药事法中,对于获得承认的产品,允许承继者继承其地位,而在医药品医疗器械等法中,认证产品的承继也成为可能。
对医疗器械贩卖或贷与业的规制
除有偿赁贷外,无偿重复连续贷与也要取得许可或备案,名称也由赁贷业改为贷与业。
咨询有关医疗器械的许可、认可及承认等相关事宜,请联系我们。
我们向新进入医疗器械行业的企业,以及现有的制造商、生产商和经销商提供与医药品医疗器械等法相关的申请支持和咨询服务。
医疗器械的申请具有高度专业性,能够完美处理这些业务的行政书士很少。
而我们提供医疗器械相关的法律服务的基础是我们作为企业代理人向日本行政机关申请与斡旋沟通时所积累的丰富经验和优秀业绩。
我们的团队拥有在全国各都道府县的申请经验。请务必与我们联系以获取更多信息。

